 SĐT:
SĐT: 
I. LỜI MỞ
Mấy ngày nay, dư luận xôn xao bàn tán về thuốc điều trị ung thư Pembrosia do Nga sản xuất vừa được Vụ Quản lý Dược Bộ y tế cấp phép nhập về dưới tên gọi là thuốc sinh học tương đương (biosimilar, generic).
Hai câu hỏi đặt ra: (1) Thuốc tương đương là gì? phát triển thế nào? và (2) Pembrosia đã hoàn chỉnh để sử dụng rộng rãi, hay còn thử nghiệm lâm sàng?
II. QUY TRÌNH PHÁT TRIỂN THUỐC SINH HỌC TƯƠNG ĐƯƠNG
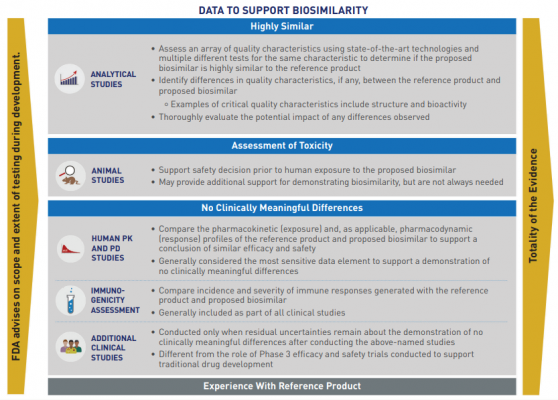
Các cơ quan quản lý dực yêu cầu thuốc sinh học tương tự không có sự khác biệt lâm sàng và phải đáp ứng tiêu chuẩn khoa học nghiêm ngặt về tính tương đồng trước khi được cung cấp cho bệnh nhân. Tính tương đồng phải qua một quy trình khoa học xác nhận không có sự khác biệt có ý nghĩa lâm sàng giữa thuốc sinh học tương tự và sản phẩm gốc. Thử nghiệm tính tương đồng được thực hiện nhiều lần trong suốt quá trình phát triển.
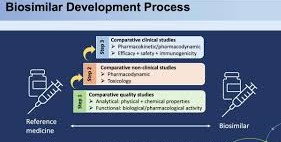
Quá trình phát triển thuốc sinh học tương tự trải qua ba giai đoạn chính: mô tả đặc tính và hoàn thiện quy trình, xác nhận tính tương đồng sinh học và phê duyệt.
1. Phát triển sản phẩm: Đặc tính và Quy trình
Trong quá trình phát triển, bước đầu tiên là hiểu rõ về thuốc sinh học tham chiếu, được thực hiện thông qua việc kiểm tra cấu trúc và chức năng. Quá trình này được gọi là đặc tính hóa.
Sau khi có được thông tin này, giai đoạn tiếp theo là phát triển quy trình sản xuất, mang lại liệu pháp có độ tương đồng cao. Các công nghệ phát triển sinh học tiên tiến và các công cụ phân tích có độ nhạy cao được sử dụng để thiết kế một cách có hệ thống một phân tử tương tự sinh học phù hợp với các thuộc tính chất lượng của thuốc đã được xác định trong giai đoạn đặc tính hóa. Đây là một quy trình lặp đi lặp lại, trong đó mỗi phần của quy trình sản xuất được tối ưu hóa theo các bước lặp lại. Quá trình này tiếp tục cho đến khi quy trình sản xuất tạo ra một cấu trúc phân tử có độ tương đồng cao với thuốc tham chiếu một cách nhất quán.
2. Xác nhận thuốc thông qua các nghiên cứu và hợp tác quản lý
Sau khi xác định được sự tương đồng giữa thuốc sinh học tương tự và thuốc sinh học tham chiếu thông qua phân tích và thử nghiệm, bước tiếp theo sẽ bắt đầu. Cơ quan quản lý dược sẽ xem xét tất cả thông tin và xác định các nghiên cứu tiền lâm sàng và lâm sàng bổ sung, nếu có, cần thiết để xác nhận tính tương đồng sinh học và khả năng hoán đổi.
Thử nghiệm lâm sàng thường được yêu cầu để phê duyệt thuốc sinh học tương tự tại các thị trường được quản lý chặt chẽ, chẳng hạn như Châu Âu, Hoa Kỳ, Nhật Bản, Canada và Úc. Tuy nhiên, phạm vi và các yêu cầu đối với các thử nghiệm lâm sàng thuốc sinh học tương tự sẽ phụ thuộc vào dữ liệu được nộp. Ví dụ, khi có dữ liệu phân tích mạnh mẽ và thuyết phục, và cần thêm dữ liệu, một chương trình thử nghiệm lâm sàng được thiết kế riêng có thể cung cấp một cách hiệu quả hơn để chứng minh tính tương đồng sinh học và khả năng hoán đổi.
3: Phê duyệt
Trong giai đoạn đầu phát triển, các nhà sản xuất thuốc sinh học tương tự sẽ gặp cơ quan quản lý dược để thảo luận về kế hoạch phát triển sản phẩm và các phương pháp cung cấp luận cứ khoa học đầy đủ trong suốt quá trình đánh giá. Trước khi phê duyệt bất kỳ sản phẩm nào cho bệnh nhân sử dụng, cơ quan quản lý dược sẽ xem xét toàn bộ bằng chứng và tiến hành đánh giá nghiêm ngặt tất cả dữ liệu để xác định xem các tiêu chuẩn khoa học hiện hành có được đáp ứng thành công hay không. Sau khi một thuốc sinh học tương tự được phê duyệt, nó có thể được sản xuất và phân phối.
III. LỜI BÀN
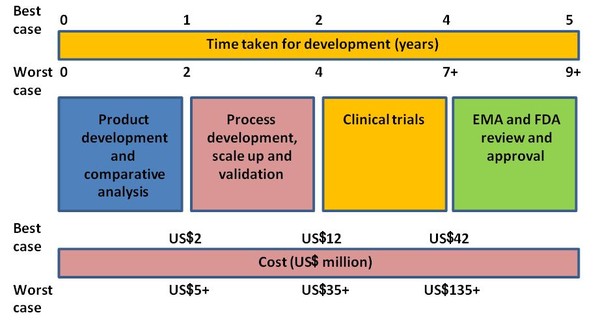
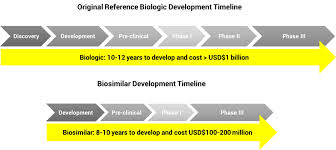
Nếu được phát triển đúng quy trình chuẩn mực nêu trên, bệnh nhân vẫn đạt được an toàn và hiệu quả gần như như các sản phẩm sinh học gốc trong khi quy trình sản xuất ngắn hơn (bỏ qua các giai đoạn các giai đoạn ý tưởng, thứ tự nghiên cứu…. ) nên giả rẻ dễ chấp nhận hơn.
Ở các nước tiên tiến, Hội đồng Thuốc sinh học tương tự (Biosimilars Council) thường nỗ lực thúc đẩy tạo điều kiện cho sự phát triển thuốc, mở đường cho luật pháp cũng như cung cấp về thuốc sinh học tương tự cho bệnh nhân.
Thắc mắc của nhiều người dân về thuốc Pembrosia mới cho nhập là có cơ sở, khi chúng ta đọc kỹ Điều 2.5 của Quyết định số 628 QĐ- QLD ngày 31/10/2025 của Cục Quản lý Dược Bộ Y tế là cho phép lưu hành để thử nghiệm lâm sàng chứ không phải là cấp phép lưu hành rộng rãi trên thị trường [1].
2.5. Thuốc số thứ tự 1 tại Phụ lục Quyết định này (Pembrosia): sau khi được cấp giấy đăng ký lưu hành thuốc, công ty phải định kỳ 03 tháng một lần kể từ ngày cấp giấy đăng ký lưu hành thực hiện cập nhật tiến độ triển khai nghiên cứu lâm sàng theo dõi tính sinh miễn dịch pha III và nộp hồ sơ thay đổi, bổ sung cập nhật dữ liệu theo dõi tính sinh miễn dịch pha III khi thời gian nghiên cứu kết thúc. [1]

IV. THAM KHẢO
[Video] Biosimilars: Totality of the Evidence in Biosimilar Development
[1] QUYẾT ĐỊNH VỀ VIỆC BAN HÀNH DANH MỤC 14 VẮC XIN, SINH PHẨM ĐƯỢC CẤP GIẤY ĐĂNG KÝ LƯU HÀNH TẠI VIỆT NAM – ĐỢT 57
https://thuvienphapluat.vn/van-ban/The-thao-Y-te/Quyet-dinh-628-QD-QLD-2025-Danh-muc-14-vac-xin-duoc-cap-giay-luu-hanh-tai-Viet-Nam-Dot-57-679345.aspx
[2] How a Biosimilar is Developed
[3] Understanding the Development and Manufacturing Process of Biosimilars
https://www.susupport.com/blogs/knowledge/understanding-development-and-manufacturing-process-biosimilars
[4] Biosimilar Development Process
https://www.fda.gov/files/drugs/published/Biosimilar-Development-Process.pdf
[5] Steps of manufacturing a biosimilar
https://www.gabionline.net/biosimilars/general/Amgen-explains-the-steps-of-manufacturing-a-biosimilar
[6] The development of biosimilars
https://www.pfizerbiosimilars.com/biosimilars-development
[7] The Art of Biosimilar Reverse Engineering: From Complex Molecules to Therapeutic Equivalence
The Art of Biosimilar Reverse Engineering: From Complex Molecules to Therapeutic Equivalence
TS.BS Trần Bá Thoại
Ủy viên BCH Hội NỘI TIẾT VIỆT NAM